User guide
Note
If you are just starting to use WhatsHap, we recommend that you read our book chapter Read-Based Phasing and Analysis of Phased Variants with WhatsHap, which is a more recent and concise introduction than this guide.
WhatsHap is a read-based phasing tool. In the typical case, it expects 1) a VCF file with variants of an individual and 2) a BAM or CRAM file with sequencing reads from that same individual. WhatsHap uses the sequencing reads to reconstruct the haplotypes and then writes out the input VCF augmented with phasing information.
The basic command-line for running WhatsHap is this:
whatshap phase -o phased.vcf --reference=reference.fasta input.vcf input.bam
The reads used for variant calling (to create the input VCF) do not need to be the same as the ones that are used for phasing. We recommend that high-quality short reads are used for variant calling and that the phasing is then done with long reads, see the recommended workflow.
If the input VCF is a multi-sample VCF, WhatsHap will haplotype all samples individually. For this, the input must contain reads from all samples.
Multiple BAM/CRAM files can be provided, even from different technologies.
If you want to phase samples of individuals that are related, you can use pedigree phasing mode to improve results. In this mode, WhatsHap is no longer purely a read-based phasing tool.
You can also phase indels by adding the option --indels.
Providing a FASTA reference with --reference is highly recommended, in
particular for error-prone reads (PacBio, Nanopore), as it is enables the
re-alignment variant detection algorithm. If a reference is not available,
--no-reference can instead be provided, but at the expense of phasing
quality.
WhatsHap adds the phasing information to the input VCF file and writes it to the output VCF file. See below to understand how phasing information is represented.
The VCF file can also be gzip-compressed.
Features and limitations
WhatsHap can phase SNVs (single-nucleotide variants), insertions, deletions, MNPs (multiple adjacent SNVs) and “complex” variants. Complex variants are those that do not fall in any of the other categories, but are not structural variants. An example is the variant TGCA → AAC. Structural variants are not phased.
If no reference sequence is provided (using --reference), only
SNVs, insertions and deletions can be phased.
All variants in the input VCF that are marked as being heterozygous (genotype 0/1) and that have appropriate coverage are used as input for the core phasing algorithm. If the algorithm could determine how the variant should be phased, that information will be added to the variant in the output VCF.
Variants can be left unphased for two reasons: Either the variant type is not supported or the phasing algorithm could not make a phasing decision. In both cases, the information from the input VCF is simply copied to the output VCF unchanged.
Subcommands
WhatsHap comes with the following subcommands.
Subcommand |
Description |
|---|---|
phase |
Phase diploid variants |
Phase polyploid variants |
|
Phase polyploid variants |
|
Print phasing statistics |
|
Compare two or more phasings |
|
hapcut2vcf |
Convert hapCUT output format to VCF |
unphase |
Remove phasing information from a VCF file |
Tag reads by haplotype |
|
Genotype variants |
|
Split reads by haplotype |
|
Generate sequencing technology specific error profiles |
|
Phase VCF file using haplotagged BAM file |
Not all are fully documented in this manual, yet. To get help for a
subcommand named SUBCOMMAND, run
whatshap SUBCOMMAND --help
Recommended workflow
Best phasing results are obtained if you sequence your sample(s) on both PacBio and Illumina: Illumina for high-quality variant calls and PacBio for its long reads.
1. Map your reads to the reference, making sure that you assign each read to a
read group (the @RG header line in the BAM/CRAM file). WhatsHap supports VCF
files with multiple samples and in order to determine which reads belong to which
sample, it uses the ‘sample name’ (SM) of the read group. If you have a single
sample only and no or incorrect read group headers, you can run WhatsHap with
--ignore-read-groups instead.
2. Call variants in your sample(s) using the most accurate reads you have. These will typically be Illumina reads, resulting in a a set of variant calls you can be reasonably confident in. If you do not know which variant caller to use, yet, we recommend FreeBayes, which is fast, Open Source and easy to use. In any case, you will need a standard VCF file as input for WhatsHap in the next step.
3. Run WhatsHap with the VCF file of high-confidence variant calls (obtained in the previous step) and with the longest reads you have. These will typically be PacBio reads. Phasing works best with long reads, but WhatsHap can use any read that covers at least two heterozygous variant calls, so even paired-end or mate-pair reads are somewhat helpful. If you have multiple sets of reads, you can combine them by providing multiple BAM/CRAM files on the command line.
Input data requirements
WhatsHap needs correct metadata in the VCF and the BAM/CRAM input files so that it can figure out which read belongs to which sample. As an example, assume you give WhatsHap a VCF file that starts like this:
##fileformat=VCFv4.1
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT SampleA SampleB
chr1 100 . A T 50.0 . . GT 0/1 0/1
...
WhatsHap sees that there are two samples in it named “SampleA” and “SampleB”
and expects to find the reads for these samples somewhere in the BAM/CRAM file
(or files) that you provide. For that to happen, all reads belonging to a sample
must have the RG tag, and at the same time, the read group must occur in the
header of the BAM/CRAM file and have the correct sample name. In this example, a
header might look like this:
@HD VN:1.4 SO:coordinate
@SQ SN:... LN:...
...
@RG ID:1 SM:SampleA
@RG ID:2 SM:SampleB
The @RG header line will often contain more fields, such as PL for
the platform and LB for the library name. WhatsHap only uses the SM
attribute.
With the above header, the individual alignments in the file will be tagged with
a read group of 1 or 2. For example, an alignment in the BAM/CRAM file
that comes from SampleA would be tagged with RG:Z:1. This is also described
in the SAM/BAM specification.
It is perfectly fine to have multiple read groups for a single sample:
@RG ID:1a SM:SampleA
@RG ID:1b SM:SampleA
@RG ID:2 SM:SampleB
What to do when the metadata is not correct
If WhatsHap complains that it cannot find the reads for a sample, then chances are that the metadata in the BAM/CRAM and/or VCF file are incorrect. You have the following options:
Edit the sample names in the VCF header.
Set the correct read group info in the BAM/CRAM file, for example with the Picard tool AddOrReplaceReadGroups.
Re-map the reads and pass the correct metadata-setting options to your mapping tool.
Use the
--ignore-read-groupsoption of WhatsHap. In this case, WhatsHap ignores all read group metadata in the BAM/CRAM input file(s) and assumes that all reads come from the sample that you want to phase. In this mode, you can only phase a single sample at a time. If the input VCF file contains more than one sample, you need to specify which one to phase by using--sample=The_Sample_Name.
Using multiple input BAM/CRAM files
WhatsHap supports reading from multiple BAM or CRAM files. Just provide all BAM and CRAM files you want to use on the command-line. All the reads across all those files that to a specific sample are used to phase that sample. This can be used to combine reads from multiple technologies. For example, if you have Nanopore reads in one BAM file and PacBio reads in another CRAM file, you can run the phasing like this:
whatshap phase -o phased.vcf --reference=reference.fasta input.vcf nanopore.bam pacbio.cram
You need to make sure that read group information is accurate in all files.
Using a phased VCF instead of a BAM/CRAM file
It is possible to provide a phased VCF file instead of a BAM/CRAM file. WhatsHap will then treat the haplotype blocks (phase sets) it describes as “reads”. For example, if the phased VCF contains only chromosome-sized haplotypes, then each chromosome would give rise to two such “reads”. These reads are then used as any other read in the phasing algorithm, that is, they are combined with the normal sequencing reads and the best solution taking all reads into account is computed.
Read selection and merging
Whatshap has multiple ways to reduce the coverage of the input —
allowing faster runtimes — in a way that attempts to minimize the
amount of information lost in this process. The default behaviour is
to ensure a maximum coverage via read selection: a heuristic that
extracts a subset of the reads that is most informative for phasing.
An optional step which can be done before selection is to merge
subsets of reads together to form superreads according to a
probabilistic model of how likely subsets of reads are to appear
together on the same haplotype (p_s) or different haplotypes (p_d).
By default, this feature is not activated, however it can be activated
by specifying the --merge-reads flag when running whatshap
phase. This model is parameterized by the following four parameters
Parameter |
Description |
|---|---|
error-rate |
Probability that a nucleotide is wrong |
maximum-error-rate |
Maximum error any edge of the merging graph can have |
threshold |
Threshold ratio of p_s/p_d to merge two sets |
negative-threshold |
Threshold ratio of p_d/p_s to not merge two sets |
which can be specified by the respective flags --error-rate=0.15,
--maximum-error-rate=0.25, --threshold=100000 and
--negative-threshold=1000 (note that defaults are shown here for
example) when running whatshap phase.
Representation of phasing information in VCFs
WhatsHap supports two ways in which it can store phasing information in a VCF
file: The standards-compliant PS tag and the HP tag used by GATK’s
ReadBackedPhasing tool. When you run whatshap phase, you can select which
format is used by setting --tag=PS or --tag=HP.
We will use a small VCF file as an example in the following. Unphased, it looks like this:
##fileformat=VCFv4.1
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT sample1 sample2
chr1 100 . A T 50.0 . . GT 0/1 0/1
chr1 150 . C G 50.0 . . GT 0/1 1/1
chr1 300 . G T 50.0 . . GT 0/1 0/1
chr1 350 . T A 50.0 . . GT 0/1 0/1
chr1 500 . A G 50.0 . . GT 0/1 1/1
Note that sample 1 is heterozygous at all shown loci (expressed with
0/1 in the GT field).
Phasing represented by pipe (|) notation
The GT fields can be phased by ordering the alleles by haplotype and
separating them with a pipe symbol (|) instead of a slash (/):
##fileformat=VCFv4.1
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT sample1 sample2
chr1 100 . A T 50.0 . . GT 0|1 0/1
chr1 150 . C G 50.0 . . GT 1|0 0/1
chr1 300 . G T 50.0 . . GT 1|0 0/1
chr1 350 . T A 50.0 . . GT 0|1 0/1
chr1 500 . A G 50.0 . . GT 0|1 1/1
The alleles on one of the haplotypes of sample1 are: A, G, T, T, A. On the other haplotype, they are: T, C, G, A, G.
Swapping ones and zeros in the GT fields would result in a VCF file with
the equivalent information.
Phasing represented by PS (“phase set”) tag
The pipe notation has problems when not all variants in the VCF file can be
phased. The VCF specification
introduces the PS tag to solve some of them. The PS is a
unique identifier for a “phase set”, which is a set of variants that were
be phased relative to each other. There are usually multiple phase sets in
the file, and variants that belong to the same phase set do not need to
be consecutive in the file:
##fileformat=VCFv4.1
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT sample1 sample2
chr1 100 . A T 50.0 . . GT:PS:PQ 0|1:100:22 0/1:.:.
chr1 150 . C G 50.0 . . GT:PS:PQ 1|0:100:18 0/1:.:.
chr1 300 . G T 50.0 . . GT:PS:PQ 1|0:300:23 0/1:.:.
chr1 350 . T A 50.0 . . GT:PS:PQ 0|1:300:42 0/1:.:.
chr1 500 . A G 50.0 . . GT:PS:PQ 0|1:100:12 0/1:.:.
This VCF contains two phase sets named 100 and 300. The names are
arbitrary, but WhatsHap will choose the position of the leftmost variant
of the phase set as its name. The variants at 100, 150 and 500 are in the same
phase set, while the variants at 300 and 350 are in a different phase set.
Such a configuration is typically seen when paired-end or mate-pair reads are
used for phasing.
In the case of WhatsHap, the phase sets are identical to the connected components of the variant connectivity graph. Two variants in that graph are connected if a read exists that covers them.
The above example also shows usage of the PQ tag for “phasing quality”.
WhatsHap currently does not add this tag.
Phasing represented by HP tag
GATK’s ReadBackedPhasing tool uses a different way to represent phased variants.
It is in principle the same as the combination of pipe notation with the PS
tag, but the GT field is left unchanged and all information is added to a
separate HP tag (“haplotype identifier”) instead. This file encodes the same
information as the example above:
##fileformat=VCFv4.1
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT sample1 sample2
chr1 100 . A T 50.0 . . GT:HP 0/1:100-1,100-2 0/1:.:.
chr1 150 . C G 50.0 . . GT:HP:PQ 0/1:100-2,100-1:18 0/1:.:.
chr1 300 . G T 50.0 . . GT:HP:PQ 0/1:300-2,300-1:23 0/1:.:.
chr1 350 . T A 50.0 . . GT:HP:PQ 0/1:300-1,300-2:42 0/1:.:.
chr1 500 . A G 50.0 . . GT:HP:PQ 0/1:100-1,100-2:12 0/1:.:.
A few notes:
ReadBackedPhasing does not add the
PQto the first variant in a phase set/haplotype group. This probably means that the phasing quality is to be interpreted as relative to the previous or first variant in the set.ReadBackedPhasing does not phase indels
- Discussions on the GATK forum on this topic:
Trusting the variant caller
WhatsHap will trust the variant caller to have made the right decision of
whether a variant is heterozygous or homozygous. If you use the option
--distrust-genotypes, then this assumption is softened: An optimal solution
could involve switching a variant from being heterozygous to homozygous.
Currently, if that option is enabled and such a switch occurs, the variant
will simply appear as being unphased. No change of the genotype in the VCF is
done.
If you use this option, fewer variants will be phased.
Note that switching homozygous variants to heterozygous is never possible since only heterozygous variants are considered for phasing.
Phasing pedigrees
When phasing multiple samples from individuals that are related (such as
parent/child or a trio), then it is possible to provide WhatsHap with
a .ped file that describes the pedigree. WhatsHap will use the
pedigree and the reads to infer a combined, much better phasing.
To turn on pedigree mode, run WhatsHap like this:
whatshap phase --ped pedigree.ped --reference=reference.fasta -o phased.vcf input.vcf input.bam
where pedigree.ped is a plink-compatible PED file to describe the
relationships between samples and input.vcf is a multi-sample VCF
with all individuals that should be phased. The reads for all individuals
can be in one or more BAM/CRAM files. WhatsHap will match them based on sample
names provided in the read groups (just like for the default single-individual
mode).
In the resulting VCF file (phased.vcf),
haplotype alleles of a child are given as paternal|maternal, i.e.
the first allele is the one inherited from the father and the second one
the allele inherited from the mother.
PED file format
WhatsHap recognizes PLINK-compatible PED files. A PED file is a white-space (space or tab) delimited file with at least six columns. WhatsHap checks the column count, but uses only
column 2: individual ID
column 3: paternal ID
column 4: maternal ID
The other columns are ignored. Lines starting with # are considered
comments and are ignored. Empty lines are also ignored.
To define a single trio, it is sufficient to have a single row in the PED file with the child, mother and father. It is not necessary to include “dummy” rows for individuals whose parents are unknown. (You will currently get a warning if you do, but this will be changed.)
Here is an example defining a trio:
# Fields: family, individual_id, paternal_id, maternal_id, sex, phenotype
FAMILY01 the_child father mother 0 1
A quartet (note how multiple consecutive spaces are fine):
# Fields: family, individual_id, paternal_id, maternal_id, sex, phenotype
FAMILY01 one_child father mother 0 1
FAMILY01 other_child father mother 0 1
Important: The names in the PED file must match the sample names in your VCF and BAM/CRAM files!
Pedigree phasing parameters
Phasing in pedigree mode requires costs for recombination events. Per
default, WhatsHap will assume a constant recombination rate across the
chromosome to be phased. The recombination rate (in cM/Mb) can be
changed by providing option --recombrate. The default value of
1.26 cM/Mb is suitable for human genomes.
In order to use region-specific recombination rates, a genetic map file
can be provided via option --genmap. WhatsHap expects a three-column
text file like this:
position COMBINED_rate(cM/Mb) Genetic_Map(cM)
55550 0 0
568322 0 0
568527 0 0
721290 2.685807669 0.410292036939447
723819 2.8222713027 0.417429561063975
723891 2.9813105581 0.417644215424158
...
The first (header) line is ignored and the three columns are expected to
give the pysical position (in bp), the local recombination rate between the
given position and the position given in the previous row (in cM/Mb), and
the cumulative genetic distance from the start of the chromosome (in cM).
The above example was taken from the 1000 Genomes genetic map provided by
SHAPEIT.
Since genetic map files provide information for only one chromosome, the
--genmap option has to be combined with --chromosome.
Creating phased references in FASTA format
To reconstruct the two haplotypes that a phased VCF describes, the
bcftools consensus command can be used. It is part of
bcftools. As input, it expects a reference
FASTA file and either an indexed BCF or a compressed and indexed VCF file.
To work with the uncompressed VCF output that WhatsHap produces, proceed
as follows:
bgzip phased.vcf
tabix phased.vcf.gz
bcftools consensus -H 1 -f reference.fasta phased.vcf.gz > haplotype1.fasta
bcftools consensus -H 2 -f reference.fasta phased.vcf.gz > haplotype2.fasta
Here, reference.fasta is the reference in FASTA format and phased.vcf
is the phased VCF. Afterwards, haplotype1.fasta and haplotype2.fasta
will contain the two haplotypes.
whatshap stats: Computing phasing statistics
The stats subcommand prints phasing statistics for a single VCF file:
whatshap stats input.vcf
The TSV statistics format
With --tsv=FILENAME, statistics are written in tab-separated value format
to a file. If you use MultiQC, the
file is automatically found and parsed and the key statistics are included in
its generated report.
The following columns are included in the TSV file.
- sample
The name of the sample the numbers in this row refer to.
- chromosome
The name of the chromosome the numbers in this row refer to. The special name “ALL” is used for summary statistics about all processed chromosomes.
- file_name
The VCF file name to which the numbers in this row refer to.
The numbers in these following columns are computed on the variant level.
- variants
Number of biallelic variants in the input VCF excluding duplicate positions and, if
--only-snvswas used, also excluding any non-SNV variants.- heterozygous_variants
The number of biallelic, heterozygous variants in the input VCF. This is a subset of variants as defined above.
- heterozygous_snvs
The number of biallelic, heterozygous SNVs in the input VCF. This is a subset of heterozygous_variants.
- unphased
The number of biallelic, heterozygous variants that are not marked as phased in the input VCF. This is also a subset of heterozygous_variants.
- phased
The number of biallelic, heterozygous variants that are marked as phased in the input VCF, excluding singletons. This is again a subset of heterozygous_variants. Add singletons to get the total number of variants marked as phased in the VCF. Also note that the following is true: phased + unphased + singletons = heterozygous_variants.
- phased_snvs
The number of biallelic, heterozygous SNVs that are marked as phased in the input VCF. This is a subset of phased.
- phased_fraction
The fraction of heterozygous variants that are phased. Same as phased / heterozygous_variants.
- phased_snvs_fraction
The fraction of heterozygous SNVs that are phased. Same as phased_snvs / heterozygous_snvs.
Each phased variant is part of exactly one phase set (stored in the PS tag in VCF) or block. The numbers in the following columns describe these blocks.
- blocks
The total number of phase sets/blocks.
- singletons
The number of blocks that contain exactly one variant.
These columns describe the distribution of non-singleton block sizes, where the size of a block is the number of variants it contains.
- variant_per_block_median
Median number of variants.
- variant_per_block_avg
Average (mean) number of variants.
- variant_per_block_min
Minimum number of variants.
- variant_per_block_max
Maximum number of variants.
- variant_per_block_sum
Sum of the number of variants. Note that this value should be the same as phased.
The following columns describe the distribution of non-singleton block lengths, where the length of a block is the number of basepairs it covers minus 1. That is, a block with two variants at positions 2 and 5 has length 3. Interleaved blocks are cut in order to avoid artificially inflating this value.
- bp_per_block_median
Median block length.
- bp_per_block_avg
Average (mean) block length.
- bp_per_block_min
Minimum block length.
- bp_per_block_max
Maximum block length.
- bp_per_block_sum
Total sum of block lengths.
- block_n50
The NG50 value of the distribution of the block lengths.
Note that this is an “NG50” (not “N50”), that is, the threshold of 50% is relative to the true length of the contig as reported in the VCF header. (For an N50, the length would be the sum of the length of all blocks). It is thus possible that the sum of all block lengths does not reach 50% of the length of the contig. In this case, the value in this column is set to 0.
If no contig lengths are available, this is set to
nan. Use--chr-lengthsto provide an external table with contig lengths in case the VCF header does not contain this information.
Writing haplotype blocks in TSV format
With option --block-list=filename.tsv, a file in tab-separated value
format (TSV) is created with the haplotype blocks, one block per line.
The columns are:
sample, chromosome, phase_set, from, to, variants.
- phase_set
value of the PS tag of this block
- from
1-based starting position of the leftmost variant in this block
- to
1-based starting position of the rightmost variant in this block
- variants
Number of variants in this block
This output format does not allow you to see interleaved haplotype blocks. Use –gtf` instead if you need this information.
As an example, assume the input is this VCF:
#CHROM POS ID REF ALT ... FORMAT sample
ref 2 . A C ... GT 0|1
ref 5 . G T ... GT 1|0
Then this will be the output:
#sample chromosome phase_set from to variants
sample ref 0 2 5 2
Writing haplotype blocks in GTF format
With --gtf=filename.gtf, a GTF file is created that describes the haplotype blocks,
see GTF with haplotype blocks.
Visualizing phasing results
Sometimes it is helpful to visually inspect phasing results by looking at them in a genome browser. The steps here assume that you use the Integrative Genomics Viewer (IGV).
GTF with haplotype blocks
WhatsHap can create a GTF file from a phased VCF file that describes the
haplotype blocks. With phasing results in phased.vcf, run
whatshap stats --gtf=phased.gtf phased.vcf
WhatsHap will print some statistics about the phasing in the VCF, and it
will also create the file phased.gtf.
Open both phased.vcf and phased.gtf in IGV in order to inspect the
haplotype block structure. In this example, there are four haplotype blocks and
it is clear which variants they connect:
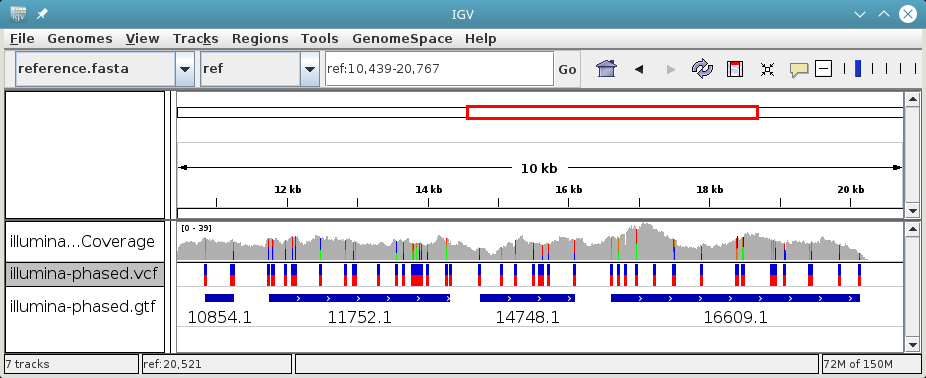
Haplotype blocks can be interleaved or nested if mate-pair or paired-end reads are used for phasing. In the GTF track, you will note this because the blocks appear as “exons” (thick segments) connected by thinner horizontal lines (not shown in the screenshot).
whatshap haplotag: Tagging reads by haplotype
If you already have a phased VCF and would like to know which reads in an
alignment file belong to which haplotype, you can use whatshap haplotag.
The tagged reads can then, for example, be visualized in IGV along with the
variants (see below).
The whatshap haplotag subcommand needs to be run on a phased VCF file.
The command tags each read in the input alignment file with
HP:i:1, HP:i:2 etc. depending on which
haplotype it belongs to, and it also adds a PS tag that describes in which
haplotype block the read is. With the aligned reads in alignments.bam,
run
whatshap haplotag -o haplotagged.bam --reference reference.fasta phased.vcf.gz alignments.bam
Add --output-threads=N with N greater than 1 to use multiple threads for compressing
the BAM file, which will speed up processing significantly.
The haplotag command requires a .vcf.gz or .bcf input file
for which an index exists (use tabix to create one).
The haplotag commands re-detects the alleles in the reads in the same way
the main phase command does it. Since availability of a reference influences
how this is done, if you used --reference with your phase command, you
should alse use --reference here.
When using 10X Genomics BAM files, haplotag reads the BX tags and per default
assigns reads that belong to the same read cloud to the same haplotype.
This feature can be switched off using the --ignore-linked-read flag.
The input VCF may have been phased by any program, not only WhatsHap, as long as
the phasing info is recorded with a PS or HP tag.
Also, the reads in the input BAM file do not have to be the ones that were used for phasing. That is, you can even phase using one set of reads and then assign haplotypes to an entirely different set of reads (but from the same sample).
The command above creates a BAM file haplotagged.bam with the tagged reads,
which you can open in IGV.
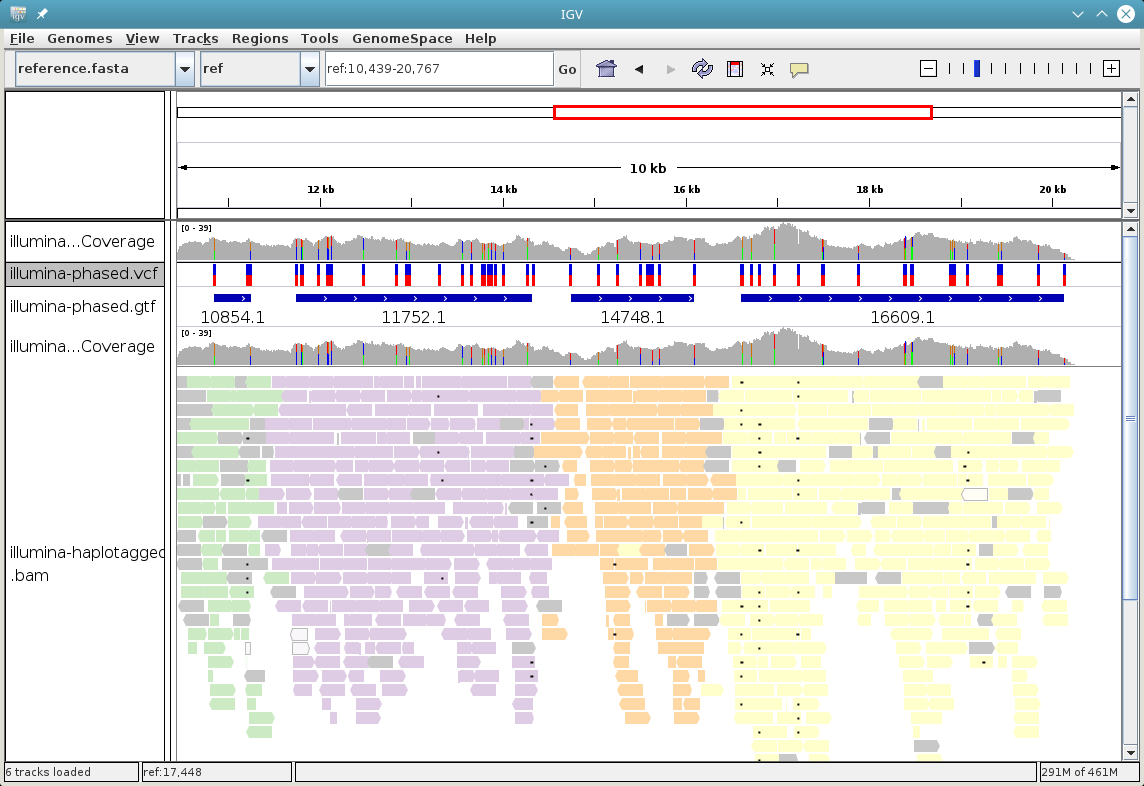
To visualize the haplotype blocks in IGV, open the BAM file in IGV,
right click on the BAM track,
and choose Color Alignments by → tag. Then type in PS and click “Ok”. Here is an
example of how this can look like. From the colors of the reads alone,
it is easy to see that there are four haplotype blocks.
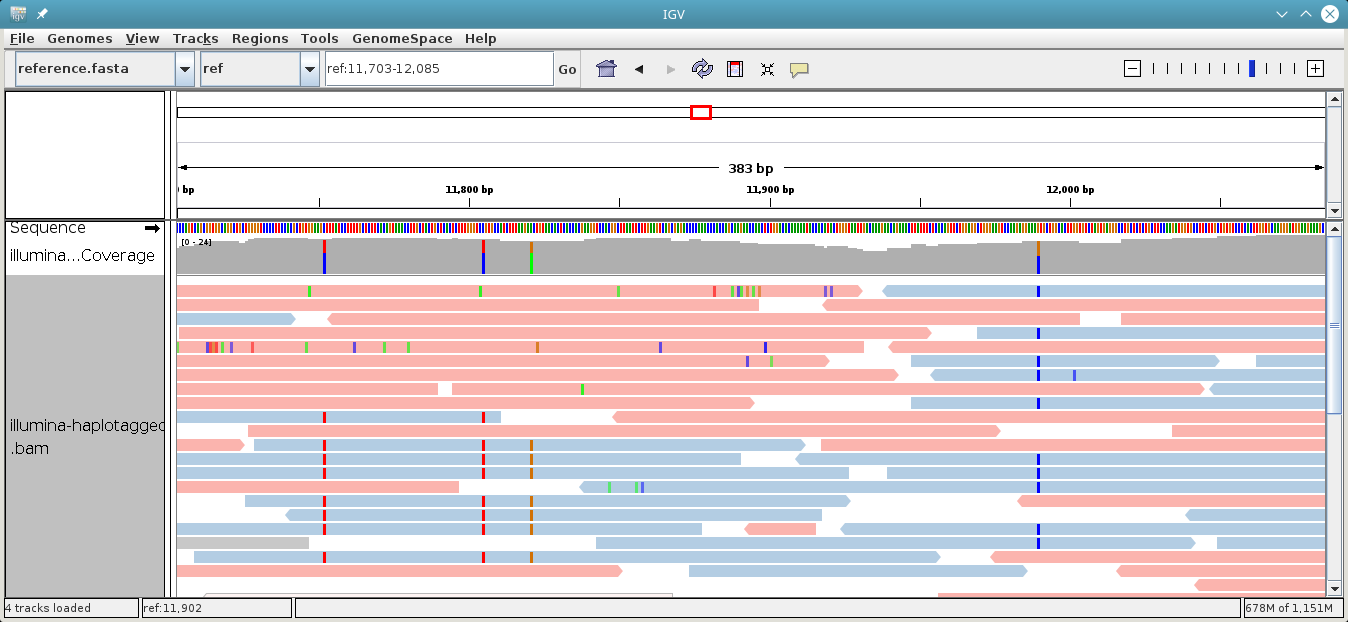
You can also visualize the haplotype assignment. For that, choose
Color Alignments by → tag and type in HP. Additionally, you may want to
also sort the alignments by the HP tag using the option Sort Alignments by
in the right-click context menu.
Here is an impression of how this can look like. The reads colored in red belong to one haplotype, while the ones in blue belong to the other. Gray reads are those that could not be tagged, usually because they don’t cover any heterozygous variants.
How haplotagging works
whatshap haplotag processes the reads in the input alignment file one by one.
(Paired-end reads are treated as a single read with a “hole” in the middle.)
If a read does not cover any heterozygous variants, nothing can be said about the haplotype, and the read remains untagged.
If a read covers at least one heterozygous variant, haplotag detects which allele(s) the
read supports. It uses the same method as whatshap phase, that is, the read
is locally re-aligned against both the reference allele and the alternative
allele, and the allele with the better alignment score wins.
Since allele detection is done separately for each variant, the detected alleles for a read do not necessarily have to agree perfectly with one of the haplotypes in the variant file. This can happen, for example, when there are sequencing errors or when the reads being haplotagged come from a different sample than the one that was used to obtain the phasing.
To resolve this, haplotag compares the alleles of the two possible
haplotypes with the alleles as found in the read. Each allele that does not
match is assigned a cost equal to the variant quality
(QUAL column in the variant file). The haplotype that incurs the
lowest sum of costs is chosen.
Finally, the HP and PS tags are added to the read accordingly.
For diploid genomes, HP can be either HP:i:1 or HP:i:2, denoting
haplotype 1 and 2.
Which haplotype is 1 and which is 2 is determined by the GT (genotype) field
in the VCF. For example consider this VCF:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT NA12878
chr1 1069577 . G A . PASS . GT:PS 1|0:1069577
The reference allele is G, the alternative allele is A.
In general, the GT field uses 0 to denote the reference allele and 1 for the
(first) alternative allele (and 2, 3, etc. for additional alternative alleles).
The | symbol in the GT field separates haplotypes, as in <haplotype1>|<haplotype2>.
Here, the first haplotype has allele 1 and the second has allele 0.
Translated to actual nucleotides, this means that haplotype 1 has allele A
and haplotype 2 has allele G.
So overall, if haplotag detects a G for that variant, it would label
the read as haplotype 2 (HP:i:2), and if it detects A, it would label
the read as haplotype 1 (HP:i:1) (assuming this is consistent with the alleles
of the other variants on the read).
Haplotagging reads with supplementary alignments
When working with long or ultra-long reads (e.g., nanopore reads) resulting alignment may contain both primary and supplementary alignments. Depending on the sample which reads are being haplotagged it is important to recognize that sometimes the data used for variants phasing (into Phase blocks and haplotypes within phase blocks). An example of such case can be working with Tumor / Normal cancer genomics. A matching normal sample N can be used for germline (heterozygous) SNP detection and phasing, producing the N.phased.vcf.gz file. Since tumor genome “inherits” all germline SNPs from the matching normal (loss is possible with LOH events), it becomes important to haplotag tumor reads based on germline SNPs and their phaseblocks/haplotype groupings. Because cancer genomes can be highly rearranged with somatic (i.e., non-germline/non-matching-normal) structural variations (e.g., translocations, long-range deletions/inversions) the reads that come from derived (rearranged) chromosomes can have both primary and supplementary alignments that can fall into rather distant regions of the genomes (even distinct chromosomes in case of translocations/chromoplexy/chromothripsis). Such primary/supplementary alignment regions may fall into phased regions (based on N.phased.vcf.gz info) with distinct phase block ids. In such cases it may beneficial to attempt to assign each alignment segment into respective phase block/haplotype based on spanned phased variants. This can help identify haplotype/ps groupings of somatic variants.
Depending on the needs of ones setup, the following strategies for treating supplementary alignments of a single read
using the --tag-supplementary CLI flag (and arguments for it enumerated below) and its values:
skip/ no flag specified – each supplementary alignment segment is skipped in assigning PS/HP SAM tagscopy-primary/ flag without argument – each supplementary alignment segment located in the same chromosome as the primary alignment segment get a copy of PS/HP SAM tags obtained for the primary alignment, if any obtained in haplotagging.independent-or-skip– each supplementary alignment is treated as an independent (i.e. as are primary alignments) case and and attempt is made to haploptag it based on spanned phased variants. If unsuccessful, no PS/HP assignment is made.independent-or-copy-primary– each supplementary alignment is treated as an independent (i.e. as are primary alignments) case and and attempt is made to haploptag it based on spanned phased variants. If unsuccessful, get a copy of PS/HP SAM tags obtained for the primary alignment, if any obtained in haplotagging and alignment segments located on the same chromosome.
When haplotagging of a supplementary has a possibility of copying a tag from teh primary alignment (e.i., copy-primary or independent-or-copy-primary) the following two flags come into play:
--supplementary-distance(default to100000) – the largest distance between closes primary and supplementary alignment positions when copy attempt is made, otherwise no supplementary tagging is performed--supplementary-strand-match(Default) /--no-supplementary-strand-match– whether the supplementary alignment needs to match in direction to the primary one for the tag copying to be performed.
whatshap split: Splitting reads according to haplotype
The whatshap split subcommand splits a set of unmapped reads from a FASTQ or BAM input file
according to their haplotype and produces one output file for each haplotype.
The haplotype for each read must be provided through a separate file, typically
created by whatshap haplotag with the --output-haplotag-list option.
This file must be in tab-separated values (TSV) format and must have at least two columns with
read name and haplotype. Two additional columns phase set and contig are required
if the command-line option --only-largest-block was used. A header line is optional.
Input reads are provided as either BAM or FASTQ. The output format is the same as the input format. That is, reading BAM but writing FASTQ (or vice versa) is not possible.
Examples:
whatshap split --output-h1 h1.fastq.gz --output-h2 h2.fastq.gz reads.fastq.gz haplotypes.tsv
whatshap split --output-h1 h1.bam --output-h2 h2.bam reads.bam haplotypes.tsv
When splitting files with ploidy greater two, an alternative syntax needs to be used:
The option -o must be provided as many times as there are haplotypes.
The first output file is used for the reads from the first haplotype,
the second for the reads from the second haplotype etc.
For example, to split a tetraploid file:
whatshap split -o h1.bam -o h2.bam -o h3.bam -o h4.bam reads.bam haplotypes.tsv
whatshap genotype: Genotyping Variants
Besides phasing them, WhatsHap can also re-genotype variants. Given a VCF file containing variant positions, it computes genotype likelihoods for all three genotypes (0/0, 0/1, 1/1) and outputs them in a VCF file together with a genotype prediction. Genotyping can be run using the following command:
whatshap genotype -o genotyped.vcf variants.vcf reads.bam
The predicted genotype is stored in the output VCF using the GT tag and the GL tag
provides (log10-scaled) likelihoods computed by the genotyping algorithm.
As for phasing, providing a reference sequence is strongly recommended in order to
enable re-alignment mode:
whatshap genotype --reference ref.fasta -o genotyped.vcf variants.vcf reads.bam
If no input VCF file is available, WhatsHap can produce candidate SNV positions that can be used as an input to the above mentioned genotyping commands. This can be done by running:
whatshap find_snv_candidates ref.fasta input.bam -o variants.vcf
If Nanopore reads are used for calling SNPs, it is recommended to add option –nanopore to the above command.
whatshap polyphase: Polyploid Phasing
In addition to diploid phasing, WhatsHap also supports polyploid phasing
through a different algorithm. The whatshap polyphase command works
similarly to the phase command:
whatshap polyphase input.vcf input.bam --ploidy p --reference ref.fasta -o output.vcf
Some details differ from the diploid command:
1. An additional integer argument --ploidy must be specified. This ploidy
must match the ploidy in the provided VCF file(s). The ploidy also greatly
impacts the running time as the phasing becomes more complex. Ploidies
higher than 6 may take very long to process.
2. WhatsHap will use available genotype information from the VCF file(s) and
output phasings that strictly follow these inputs. However, when using the
--distrust-genotypes flag, the provided genotypes will be overwritten with
what the phasing algorithm thinks is the most likely (phased) genotype.
Polyploid phasing on pedigrees is not supported yet.
4. Specifying a reference genome is optional, like for the diploid case. However, available reference genomes for plant species usually fall behind in accuracy when compared to a human one. We observed both improvements and regressions in phasing accuracy when providing the phaser with a reference sequence, so there is no clear recommendation whether to use it or not.
There is no strict limitation regarding the coverage of the input reads. However, the running time grows quadratically with the coverage, so be aware that very deep sequencing data might take a long time.
Since polyploid phasing is inherently more difficult than diploid phasing, the phased blocks are expected to be much shorter than a diploid phasing with the same input quality. A major problem are long intervals, where two or more haplotypes look (almost) completely identical. For these intervals, the output must contain the same haplotype sequence multiple times. While the multiplicity of such a sequence might be derived from allele coverages, it becomes impossible to connect the haplotype sequences before and after such an interval, unless there are sufficiently many reads that completely span the entire interval.
By default, WhatsHap will cut the phasing on such ambiguous sites. The same
applies for regions with very low heterozygosity, where only very few or even
no reads connect two consecutive variants. The parameter
--block-cut-sensitivity (or short -B) controls how conservatively the
phaser will cut the phasing. Valid values range from 0 to 5 with a default of
4. A lower sensitivity will produce longer phasing blocks, but keep in mind
that this will lead to more switch errors when haplotype sequences become
(almost) identical.
The optional flag --use-prephasing reads existing phasing information in
the input VCF and adds them to the phasing process. Unlike for the phase
command, phased blocks are not interpreted as additional reads, but as
scaffolding information to increase the continuity of phasing blocks produced
by the polyphase algorithm. Depending on the density of pre-phased variants you
might consider reducing the block cut sensitivity to lower levels.
In VCF format, it is common to specifiy the block IDs in the
Phase set identifier field (PS). Since this ID refers to the variant
itself, it is not possible to report which haplotypes should be cut and which
ones could be phased through. This information can be accessed via the HS
field in the VCF, if the --include-haploid-sets flag is set. This is a
custom field, which is only used to provide this information. It is not
supported by other tools and also the compare and stats modules of
WhatsHap will still use the common PS field to consider block borders.
WhatsHap does not support diverging ploidy for the same input files. All provided chromosomes will be assumed to follow the input ploidy. This can lead to unexpected results for organisms with different ploidies per chromosome, but also for very large deletions on one of the haplotypes.
It is possible to phase diploid samples via the polyphase command, but the
we recommend to use the phase command instead, because it uses a different
algorithm that is more specialized for the diploid case.
whatshap polyphaseg: Polyploid Phasing with progeny information
In addition to the purely read-based method, the whatshap polyphasegenetic
command runs on genotype data, derived from two parent samples and an arbitrary
number of progeny samples. The scope of this command is to phase one parent at
a time by using a high number of progeny samples with low-depth phasing
information. The Mendelian rules for allele heritage allow to determine the
co-occurence of marker alleles in the target parental sample. These marker
alleles occur, when one parent is homozygous in some allele A and the other
parent has exactly one allele that is different from A. This limits the
phasing capabilities to variants following such a pattern and to autopolyploid
species with an even ploidy. During development, it turned out that a
population of at least 50 is recommended when using an average sequencing depth
of 6 per progeny sample.
Since no read data is used here, the workflow differs from whatshap polyphase:
whatshap polyphasegenetic parent.vcf ped.txt [-P progeny.vcf] --ploidy p --sample s -o output.vcf
The parental VCF file must contain genotype information of both parent samples
and may contain genotype information for the progeny samples. If the progeny
genotypes are not present in the parental VCF, they must be provided in a
separate VCF, preceeded by the --progeny-file (or short -P) identifier.
As of now the progeny samples are required to have an AD field to provide
the allele depths per sample per variant. An example of such a file would be:
##fileformat=VCFv4.1
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT parent1 parent2 progeny1 ...
chr1 100 . A T 50.0 . . GT:AD 0/0/0/1:. 0/0/0/0:. 0/0/0/1:75,21
chr1 200 . C G,A 50.0 . . GT:AD 0/0/0/2:. 0/0/0/1:. 0/0/1/2:53,18,22
...
The relationship between the samples has to be specified in a pedigree file using three whitespace-separated columns to specify all trios to be considered:
parent_1 parent_2 progeny_1
parent_1 parent_2 progeny_2
parent_1 parent_2 progeny_3
...
The phasing output is formed from the co-occurence information of the previously described marker alleles. This yields a sparse phasing, where only selected variants are phased for the target sample, but since the continuity of the phasing is not limited by any read lengths, it will be one phasing block per chromosome.
whatshap compare: Comparing variant files
Compare truth.chr1.vcf to phased.chr1.vcf:
whatshap compare --names truth,whatshap --tsv-pairwise eval.tsv truth.chr1.vcf phased.chr1.vcf
To improve readibility, option --names is used to assign the name “truth” to the first
input file and “whatshap” to the second one. Without this option, the input files are given
names “file0”, “file1” etc.
whatshap compare asseses differences mainly in terms of switch errors,
but it also computes flip errors and Hamming distance.
For switch errors, assume there are two variant files A and B and the two phase sets have these phased genotypes:
A B
0|1 0|1
0|1 0|1
0|1 1|0
1|0 0|1
1|0 0|1
The first haplotype of file A can be written as 00011 and the first haplotype of
file B as 00100 (and the second haplotype of A as 11100 and the second of B as
11011). When counting the errors between them, whatshap compare detects one
switch error between the second and third position because the first haplotype
in A matches the first haplotye in B at positions one and two, but then the
first haplotype matches the second haplotype from position three onwards.
In other words: We can turn 00011 into 00100 by inverting all bits from position three onwards.
The Hamming distance counts the positions at which the haplotypes differ. For example, comparing 00000 to 00011 gives a Hamming distance of 2 because the haplotypes differ (in the last two alleles). On the other hand, comparing these two haplotypes incurs only one switch error.
Finally, two switch errors in a row are also counted as a flip error.
whatshap compare counts normal switch errors (which count any switches,
even those that can be seen as part of a flip error, but it also shows the
“switch/flip” decomposition, where the switches are broken down into
1) switches that are not part of a flip and 2) flip errors.
Any comparisons whatshap compare makes allow the roles of “first’ and “second” haplotype to be reversed. For example, when the first haplotype of A is 00000 and the first haplotype of B is 01111, you might guess that the Hamming distance would be 4, but that is not the case because whatshap compare notices that it is better to instead compare against the second haplotype of file B (which is 10000), resulting in Hamming distance of just 1.
Switch and flip example:
A B C
0|1 0|1 0|1
0|1 0|1 0|1
0|1 1|0 1|0
1|0 0|1 1|0
1|0 0|1 1|0
The A to B comparison contains one switch, whereas A vs C contains one flip (two switches).
Example output:
Comparing phasings for sample NA12878
FILENAMES
truth = truth.chr1.vcf
whatshap = phased.chr1.vcf
---------------- Chromosome chr1 ----------------
VARIANT COUNTS (heterozygous / all):
truth: 183135 / 314053
whatshap: 183135 / 314053
UNION: 183135 / 314053
INTERSECTION: 183135 / 314053
PAIRWISE COMPARISON: truth <--> whatshap:
common heterozygous variants: 183135
(restricting to these below)
non-singleton blocks in truth: 1
--> covered variants: 183135
non-singleton blocks in whatshap: 191
--> covered variants: 28764
non-singleton intersection blocks: 191
--> covered variants: 28764
ALL INTERSECTION BLOCKS: ---------
phased pairs of variants assessed: 28573
switch errors: 2504
switch error rate: 8.76%
switch/flip decomposition: 284/1110
switch/flip rate: 4.88%
Block-wise Hamming distance: 3365
Block-wise Hamming distance [%]: 11.70%
Different genotypes: 0
Different genotypes [%]: 0.00%
LARGEST INTERSECTION BLOCK: ---------
phased pairs of variants assessed: 1740
switch errors: 179
switch error rate: 10.29%
switch/flip decomposition: 21/79
switch/flip rate: 5.75%
Hamming distance: 505
Hamming distance [%]: 29.01%
Different genotypes: 0
Different genotypes [%]: 0.00%
The file written by --tsv-pairwise is in tab-separated values format and
has the following columns (example values are shown in parentheses).
- sample (NA12878)
Sample name as in the variant file header
- chromosome (chr1)
Chromosome name
- dataset_name0 (truth)
The name of the first dataset as specified by
--names- dataset_name1 (whatshap)
The name of the second dataset as specified by
--names- file_name0 (truth.chr1.vcf)
The file name of the first variant file
- file_name1 (phased.chr1.vcf)
The file name of the second variant file
- intersection_blocks (191)
The number of intersection blocks. Blocks of the (phase sets) of the first and second variant file are split where necessary to make them cover the same set of variants. This is the number of these smaller blocks.
covered_variants (28764)
all_assessed_pairs (28573)
- all_switches (2504)
The number of switch errors, summed up over all intersection blocks.
- all_switch_rate (0.0876)
Switch error rate of all intersection blocks. Computed as all_switches divided by all_assessed_pairs.
- all_switchflips (284/1110)
Switch/flip decomposition (sum over all intersection blocks) as nonflip_switches/flips. The first number is the number of switches that are not part of a flip; the second is the number of flip errors.
nonflip_switches + 2 * flips = all_switches
(284+2*1110 = 2504 in the example)
- all_switchflip_rate 0.0488
Switches and flips from the switch/flip decomposition added up, then divided by all_switches.
Example: (284 + 1110) / 28573 = 4.88%
blockwise_hamming (3365)
blockwise_hamming_rate (0.1170)
blockwise_diff_genotypes (0)
blockwise_diff_genotypes_rate (0.0)
largestblock_assessed_pairs (1740)
- largestblock_switches (179)
Number of switch errors in the largest intersection block.
- largestblock_switch_rate (0.1029)
Switch error rate of the largest intersection block.
- largestblock_switchflips 21/79
Switch/flip decompositon of the largest intersection block.
largestblock_switchflip_rate 0.0575
largestblock_hamming (505)
largestblock_hamming_rate (0.2901)
largestblock_diff_genotypes (0)
largestblock_diff_genotypes_rate (0.0)
het_variants0 (183135)
only_snvs (0)
Notes
whatshap compare only looks at identical variants when it compares two files. For example, if there is a variant at a position and it is A→C in one file and it is A→G in the other file (at the same position), then these are considered different variants, and they are excluded from comparisons.
whatshap learn: Generate sequencing technology specific error profiles
Given the aligned sequencing reads and a set of variants, Whatshap can be used to generate sequencing error profiles for a specific technology. It can be run using the following command:
whatshap learn reads.bam variants.vcf -r ref.fasta -k kmer_size -w window_size -o kmer_pair_counts
The kmer_pair_counts output file contains for each non-variant position in the reference genome, the observed count for each reference-read kmer pair.
A few notes:
windowspecifies the number of bases you want to ignore on each side of the variant. The default value is 25, i.e. 25 bases on the left and right side of the variant position would be ignored.It is recommended to run
whatshap learnin parallel on different chromosomes to save time, however, it is not mandatory.
k-merald
k-merald is an allele detection approach using k-mer based sequencing error profiles, and is now available as an alternative to the edit distance based allele detection in WhatsHap.
It can be used as follows:
Learn the error model
Get reference-read
kmer_pair_countsusingwhatshap learnConvert the
kmer_pair_countsinto phred-scores as follows:python3 -m whatshap.phred_scores -i kmer_pair_counts_dir -o phred_scores.txt -k kmer_size -e pseudocount_value_for_unobserved_kmer_pairs
Note that
kmer_pair_counts_diris the path to the directory containing the output from single whole genome or multiple chromosome specific iterations ofwhatshap learn.
Use
whatshap genotypewith additional arguments for k-merald based genotyping:whatshap genotype [options] --use-kmerald --reference ref.fasta variants.vcf reads.bam --kmeralign-costs phred_scores.txt --kmer-size kmer_size --kmerald-gappenalty gap_cost --kmerald-window window_size -o genotyped_variants.vcf
whatshap haplotagphase: Phase VCF file using haplotagged BAM file
Given a haplotagged BAM file, a file with variants, and a reference, this command sequence outputs a phased VCF file:
tabix input.vcf.gz
samtools index haplotagged.bam
whatshap haplotagphase [options] -r reference.fasta input.vcf.gz haplotagged.bam -o output.vcf.gz
It assigns phase information to a variant if the majority of reads containing this variant support the assignment. Additionally, it does not assign phase information to variants located within long homopolymers. The command supports the following options:
-g xAssigns information to the variant only if at least x percent of reads support the assignment (default value is 70).
-c xIgnores variants that lie inside homopolymers of length at least x (default value is 10).
--only-indelsAssigns information only to indel events.
--no-mavIgnore multiallelic variants.